CHE COS’E’ LA TALASSEMIA
La talassemia, detta anche Anemia Mediterranea, Malattia di Cooley, Beta Talassemia o Talassemia Major, è una grave forma di anemia emolitica ereditaria, quindi non contagiosa, ossia di una malattia dei globuli rossi carenti di emoglobina, esposti a una continua e rapida distruzione. Fra le malattie a carico del sangue è la più diffusa: oggi in Italia i talassemici sono circa 7.000, mentre oltre 3 milioni sono i portatori sani.
La talassemia è nota anche come anemia mediterranea a causa della distribuzione geografica: la talassemia si verifica più spesso nelle persone di origine italiana, greca, medio-orientale, sud-asiatica ed africana.
CAUSE DELLA MALATTIA
 La causa della talassemia è rappresentata dalla presenza di difetti nei geni dell’emoglobina, a livello del DNA: l’unico modo per contrarre la talassemia è ereditare uno o più geni di emoglobina difettosi dai propri genitori. L’anemia mediterranea è quindi una malattia ereditaria, trasmessa quando entrambi i genitori sono portatori del difetto (e peraltro completamente sani, per questo si definiscono “portatori sani”).
La causa della talassemia è rappresentata dalla presenza di difetti nei geni dell’emoglobina, a livello del DNA: l’unico modo per contrarre la talassemia è ereditare uno o più geni di emoglobina difettosi dai propri genitori. L’anemia mediterranea è quindi una malattia ereditaria, trasmessa quando entrambi i genitori sono portatori del difetto (e peraltro completamente sani, per questo si definiscono “portatori sani”).
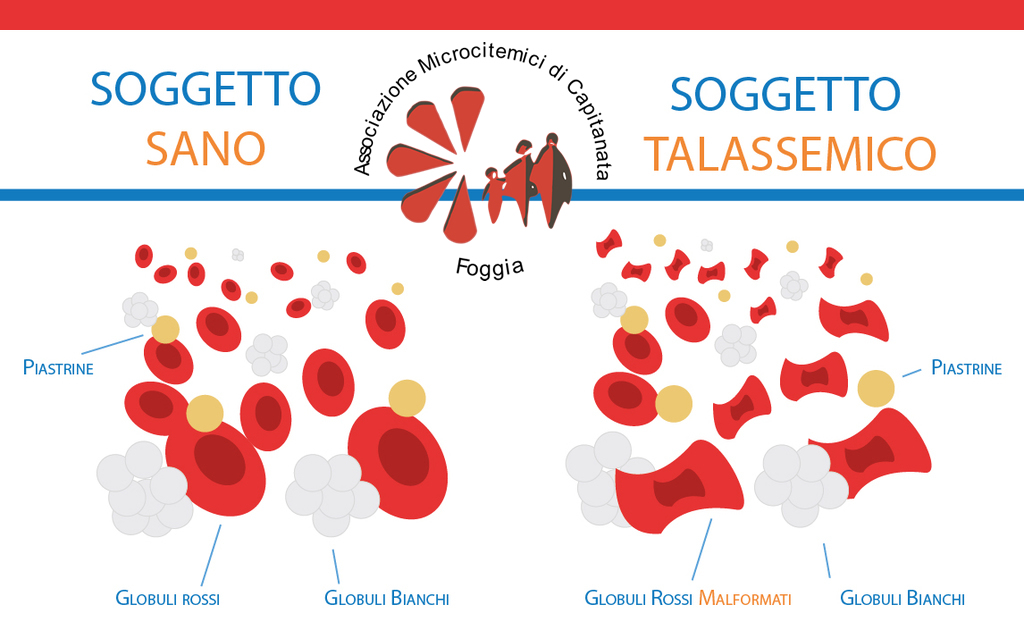
Per un’alterazione genetica, il midollo osseo del talassemico non può produrre emoglobina normale. L’emoglobina è la proteina contenuta nel globulo rosso con la vitale funzione di trasporto e scambio di ossigeno e anidride carbonica nell’organismo. L’anomalia dell’emoglobina determinerà nel sangue della persona talassemica la presenza di globuli rossi deboli e incapaci di assolvere il loro compito.
Oggi è possibile una diagnosi prenatale precoce per scoprire se il feto è affetto da anemia mediterranea con un semplice test chiamato Celocentesi. Una coppia di portatori sani ha il 25% di probabilità di generare figli talassemici e il 50% di probabilità di avere figli portatori sani. Quando uno solo dei due partner è portatore sano, il bambino potrà essere sano o nascere, a sua volta portatore sano. Si calcola che in Italia nascano 150 talassemici ogni anno.
ALFA TALASSEMIA E BETA TALASSEMIA
Ci sono due tipi principali di talassemia, alfa e beta, dal nome delle due catene proteiche dell’emoglobina che possono essere affette dall’errore genetico: in Africa è più diffusa l’alfa talassemia, mentre nel bacino del Mediterraneo è più diffusa la beta talassemia (da un punto di vista di definizioni la vera anemia mediterranea). Il tipo di anemia di cui si soffre dipende dal tipo di difetto genetico che si eredita.
Alfa Talassemia: quattro geni sono coinvolti nella sintesi della catena alfa dell’emoglobina e di questi se ne ottengono due da ciascun genitore; se uno o più geni alfa dell’emoglobina sono difettosi allora si sviluppa l’alfa-talassemia.
Se uno solo dei geni alfa dell’emoglobina è difettoso, non si hanno segni o sintomi di talassemia, ma si è comunque portatori della malattia e la si può trasmettere ai propri figli.
Se si dispone di due geni alfa dell’emoglobina difettosi, i segni ed i sintomi della talassemia sono lievi. Questo caso può essere diagnosticato come alfa-talassemia minore e si può dire che si ha un solo tratto della malattia.
Se tre dei geni alfa dell’emoglobina sono difettosi, i segni ed i sintomi della malattia possono essere da lievi a gravi. Questa condizione è anche chiamata malattia H dell’emoglobina.
Quando tutti e quattro i geni alfa dell’emoglobina sono difettosi si parla di alfa-talassemia maggiore o idrope fetale. Generalmente ciò determina la morte del feto prima del parto o subito dopo la nascita.
Beta Talassemia: due geni sono coinvolti nella sintesi della catena beta dell’emoglobina e se ne ottiene uno da ciascun genitore, se uno o entrambi i geni sono difettosi si sviluppa la beta-talassemia.
Se uno solo dei geni beta dell’emoglobina è difettoso si avvertono segni e sintomi lievi della malattia. In tal caso si parla di beta-talassemia minore o ci si riferisce ad essa come ad un tratto della beta-talassemia.
Se entrambi i geni beta dell’emoglobina sono difettosi, allora i segni ed i sintomi della malattia possono essere da moderati a gravi. In questo caso si parla di beta-talassemia maggiore o di anemia di Cooley.
 QUANDO SI MANIFESTA
QUANDO SI MANIFESTA

Intorno ai primi mesi di vita, al passaggio dalla produzione di emoglobina fetale a quella adulta. Durante lo stato fetale, infatti, nel bambino talassemico, l’emoglobina è ancora normale. Si altererà nella fase adulta, quando entrerà in attività produttiva il gene difettoso trasmesso dai genitori.
COSA VUOL DIRE ESSERE TALASSEMICI OGGI
Diagnosticata in tempo, la talassemia non impedisce un’esistenza normale. A condizioni che chi ne è affetto si sottoponga periodicamente (ogni 15 – 20 giorni) e per tutta la vita, a trasfusioni per sopperire la mancanza di globuli rossi. Inoltre, per eliminare l’eccesso di ferro che le continue trasfusioni depositano nell’organismo, è necessario che il talassemico assuma tutti i giorni, per 12 ore consecutive, un farmaco ferrochelante, per via sottocutanea, tramite un apparecchio elettronico.
IL PORTATORE SANO
 La talassemia, o anemia mediterranea, è una patologia dei globuli rossi che risultano carenti di emoglobina e sono per questo esposti a una continua e rapida distruzione.
La talassemia, o anemia mediterranea, è una patologia dei globuli rossi che risultano carenti di emoglobina e sono per questo esposti a una continua e rapida distruzione.
Due cose è opportuno specificare:
1. La Talassemia è una malattia genetica ereditaria. NON è quindi una malattia INFETTIVA!
2. Lo stato di PORTATORE SANO è una condizione di totale salute e benessere.
Chiunque potrebbe esserlo senza saperlo; per questo per verificare il proprio stato occorre eseguire uno specifico test di laboratorio in un centro accreditato e specializzato. Si tratta di un particolare esame del sangue che rileva il numero e il volume dei globuli rossi e la frazione percentuale dell’emoglobina.
Una coppia si definisce “a rischio” se i partner sono entrambi portatori sani. In questo caso si potranno verificare le seguenti combinazioni:
nel 25% dei casi il feto sarà affetto da talassemia
nel 50% dei casi il feto sarà portatore sano di talassemia (la talassemia si manifesta fin dai primi mesi di vita; il bambino è sottoposto a continue cure: trasfusioni di sangue oltre che a terapia chelante).
nel 25% dei casi il feto sarà del tutto sano.
In caso di gravidanza a rischio, oggi è possibile conoscere lo stato del feto tramite Celocentesi già dalla 7 ª settimana di gestazione. Il test di diagnosi prenatale dà risultati sicuri al 100% (così pure l’amniocentesi, eseguibile però a partire dalla 17 ª settimana).
COSA SUCCEDE SE NON SI CURA
Se non si pone alcun rimedio a questa situazione, la grave anemia che si determina diviene incompatibile con la vita: il sangue diventa privo di adeguate quantità di globuli rossi e quindi di ossigeno da distribuire a tutto l’organismo. Per continuare a vivere, questi soggetti hanno quindi bisogno di continue trasfusioni e terapie specifiche!
COSA COMPORTA LA MALATTIA

Il midollo osseo dei soggetti talassemici non è capace di produrre quantità giuste e normali di emoglobina, il pigmento che dà il colore rosso al sangue, ma che soprattutto ha il compito di trasportare l’ossigeno attraverso molecole di ferro.
I globuli rossi di questi pazienti sono pallidi, piccoli e deformati. Quando arrivano alla milza vengono riconosciuti come difettosi e precocemente distrutti: il sangue si ritrova quindi povero di globuli rossi, e il ferro viene liberato nel sangue e va a depositarsi nel pancreas, nel fegato e nel cuore. La milza continuerà la sua attività di distruzione dei globuli rossi, crescendo di dimensioni (splenomegalia). Il midollo osseo cercherà di compensare questa distruzione con una mole di lavoro superiore alla norma: aumenta quindi lo spessore di alcune ossa, soprattutto del cranio e degli zigomi, conferendo una caratteristica faccia di tipo mongoloide. Nelle ossa lunghe aumenta la componente spugnosa, che provoca osteoporosi e maggiore facilità di fratture. L’iperattività del midollo osseo, peraltro, è in buona parte inutile, perché i globuli rossi prodotti sono comunque difettosi e vengono distrutti.
Ogni globulo rosso distrutto lascia libera nel sangue una piccola quantità di ferro. A milioni di globuli rossi corrispondono proporzionali quantità di ferro, che solo in piccola parte viene eliminato: si deposita quindi in diversi organi. Ma è il cuore la sede più importante, perché la cardiopatia del talassemico, legata appunto all’accumulo di ferro, rappresenta la principale causa di mortalità. Monitorare l’accumulo cardiaco di ferro (e la corrispondente fibrosi), e seguirne l’evoluzione nel tempo e in rapporto a diverse terapie, può essere quindi determinante per sorvegliare uno dei fenomeni più deleteri nella talassemia e per individuare le scelte terapeutiche con maggiori probabilità di efficacia. La terapia fondamentale della talassemia si basa infatti su farmaci capaci di legare il ferro e di eliminarlo, prima che questo si depositi.
Le capacità intellettive di questi soggetti sono invece completamente normali. Anzi, le persone costrette a un impegno esistenziale concreto contro un serio problema di salute sviluppano spesso capacità di applicazione e concentrazione, per esempio nello studio, sensibilmente più efficaci.
COMPLICAZIONI DELLA MALATTIA
Le possibili complicanze della talassemia sono numerose e dipendono dal grado di gravità della malattia. Le complicazioni più comuni includono:
– eccessivi valori di ferro
– infezioni
– deformità ossee
– rallentamento della crescita
– ingrossamento della milza
– problemi cardiaci
QUALI SPERANZE DI GUARIGIONE
A tutt’oggi solamente il trapianto del midollo osseo può offrire una speranza di guarigione. Purtroppo questa pratica presenta diverse difficoltà, prima fra tutte il reperimento di un donatore compatibile. Basti pensare che anche tra fratelli esiste solo una possibilità su quattro di compatibilità. Per questo l’attenzione di migliaia di talassemici è rivolta alla ricerca scientifica, che ha già individuato le strade da seguire per una cura definitiva.
TALASSEMIA, UN MALE ANTICO…
L’Italia, come buona parte dei Paesi affacciati sul Mediterraneo, presentava e ha presentato fino a non moltissimo tempo fa zone paludose. In queste aree malsane, dalla Sicilia al delta padano, la malaria imperversava, risparmiando però i portatori sani di talassemia. Per questa ragione, la malattia ha potuto continuare a trasmettersi per via ereditaria, di generazione in generazione. In seguito, i flussi le hanno consentito di diffondersi in tutto il mondo. Non a caso, la Talassemia è stata individuata per la prima volta in America, nel 1925, dai pediatri Cooley e Lee su bambini greci e italiani. Ed ecco la ragione dei suoi nomi: Talassemia dal greco “thalassa”, mare, e anemia mediterranea, con chiare indicazioni dell’origine. Fortunatamente oggi di Talassemia non si muore: ben 7.000 italiani convivono con questa malattia, senza impedimenti a condurre una normale attività lavorativa, scolastica, sportiva.
…CHE LA RICERCA SA COME CURARE
I più recenti risultati della ricerca scientifica mondiale confermano che la Talassemia è un male tutt’altro che incurabile. A livello sperimentale esistono almeno due interessanti soluzioni.
Il malato di Talassemia Major o Morbo di Cooley è affetto da una grave anemia cronica sin dai primi mesi di vita.
La terapia attualmente in uso consiste in frequenti trasfusioni di sangue, mediamente ogni 15 giorni. Queste comportano un accumulo di ferro nel cuore, nel fegato e nelle ghiandole endocrine che, per essere eliminato, necessita della somministrazione di un farmaco chiamato deferrossamina, viene iniettato al soggetto talassemico sottocute, a mezzo di una pompa ad infusione lenta, per otto-dieci ore al giorno.
Nonostante i notevoli progressi della terapia convenzionale, non si può ancora parlare di guarigione definitiva dalla Talassemia e dalle emoglobinopatie.
LE TERAPIE: dalla mortalità alla sopravvivenza, verso la guarigione
L’evoluzione della terapia nel trattamento dell’anemia mediterranea ha completamente trasformato nel tempo la storia di questa malattia, portandola da una sventura rapidamente fatale a una condizione compatibile con una lunga sopravvivenza e con una qualità di vita molto buona.
Il percorso del progresso terapeutico non è completo: a breve termine i farmaci migliori, e a medio termine le terapie geniche, sono destinati a migliorare ulteriormente le prospettive di vita di questi pazienti.
La successione di risorse terapeutiche ha segnato a tappe la prognosi della talassemia.
La malattia abbandonata a se stessa
Senza cure, il paziente talassemico perde rapidamente emoglobina e globuli rossi, e il midollo osseo non è in grado di sopperire a questa continua distruzione. Senza globuli rossi ed emoglobina la fornitura di ossigeno a tutti gli organi e i tessuti dell’organismo viene progressivamente a esaurirsi, determinando una condizione incompatibile con la vita.
La malattia è quindi mortale, a distanza di settimane o mesi dalla nascita, in funzione della sua gravità.
 Le trasfusioni di sangue
Le trasfusioni di sangue
 Le trasfusioni di sangue
Le trasfusioni di sangueLa soluzione più istintiva per rimediare al problema è quella delle trasfusioni di sangue. L’immissione di globuli rossi freschi permette al paziente di avere un sangue circolante adeguato per le esigenze dell’organismo.
Il paziente talassemico esegue mediamente due trasfusioni di sangue al mese e relative visite di controllo. Ovviamente questa procedura costituisce un rimedio transitorio: i nuovi globuli rossi si legano comunque all’emoglobina alterata, prodotta dal midollo osseo, e subiscono invariabilmente il processo di distruzione precoce.
Occorrono quindi continue trasfusioni e per tutta la vita.
La somministrazione di globuli rossi giovani offre qualche vantaggio, come pure l’asportazione della milza, riducendo così l’attività distruttiva sulle cellule del sangue.
Si presenta però il problema dell’accumulo di ferro che si libera dall’emoglobina, passa nel circolo sanguigno e va a depositarsi in vari organi — soprattutto fegato, cuore, pancreas — danneggiandone inesorabilmente la funzione, e limitando la vita di queste persone a dieci-venti anni.
La perdita di funzione cardiaca, in conseguenza dell’accumulo di ferro e della fibrosi, è la principale causa di mortalità nei malati talassemici.
Terapia chelante: i farmaci leganti del ferro
Le trasfusioni creano accumuli di ferro in organi vitali. Una drastica soluzione al problema dei depositi di ferro è venuta (negli anni ’70) da farmaci capaci di legarsi a questo metallo circolante (ferrochelanti) ed eliminarlo con le urine. La desferoxamina è stata per decenni l’unico farmaco che ha consentito un cospicuo prolungamento della vita dei malati talassemici, i quali hanno comunque sempre bisogno di trasfusioni.
Questo miglioramento non è senza disagi. La somministrazione del farmaco avviene sottocute con infusione molto lenta (dodici ore) e va ripetuta cinque-sette volte alla settimana.
Non tutto il ferro viene eliminato: il problema dell’accumulo nel cuore e nel fegato viene quindi spostato nel tempo, costituendo infine la principale causa di mortalità (soprattutto cardiaca).
I nuovi ferrochelanti
A partire dagli inizi del 2000 si sono affiancati alla desferoxamina altre molecole capaci di legarsi al ferro, il deferiprone e il deferasirox, assumibili facilmente per bocca, liberando quindi dalla schiavitù dell’infusione continua.
Questi farmaci (talvolta somministrati in maniera orale) sembrano, inoltre, dotati di un’efficacia chelante maggiore. Il Deferiprone riduce l’accumulo di ferro negli organi, soprattutto nel cuore, riducendo la disfunzione cardiaca di cinque volte dopo sei anni di terapia, a parità di condizioni iniziali. Il Deferasirox è approvato per il trattamento del sovraccarico marziale in diverse anemie trasfusioni dipendenti a partire dai 2 anni di età e per il sovraccarico di ferro nelle sindromi talassemiche non trasfusioni dipendenti a partire dai 10 anni di età.
Anche se non costituiscono la soluzione definitiva, i chelanti orali contribuiscono notevolmente a migliorare la qualità della vita della persona talassemica. Come si è visto, è indispensabile che il talassemico faccia costantemente uso di un farmaco ferrochelante. Qualora non seguisse questa terapia in modo corretto, cuore e fegato ne verrebbero danneggiati. Attualmente sono disponibili due farmaci ferrochelanti somministrabili oralmente, purtroppo non da tutti ben tollerati e quindi molti dei ragazzi talassemici sono costretti tramite un microinfusore a iniettarsi quotidianamente un altro farmaco che richiede una prolungata quantità di tempo per essere introdotto nell’organismo. Quindi, questi ragazzi devono “legarsi” a questo apparecchio per dodici ore al giorno.
Il trapianto di midollo
È una tecnica con possibilità limitate. Prevede la distruzione delle cellule del midollo, e la successiva reintegrazione con cellule sane geneticamente simili (donatore compatibile). Il trapianto di midollo è ritenuta una metodica più rischiosa rispetto al rischio di mortalità naturale della malattia stessa, e quindi ormai sempre meno utilizzata.
Terapia genica
La più grossa speranza di sconfiggere la Talassemia proviene dalla bioingegneria, una scienza all’avanguardia che potrà offrire risultati risolutivi anche per le altre malattie genetiche. Da oltre dieci anni i ricercatori hanno isolato e studiato il gene responsabile della ridotta produzione di emoglobina nel talassemico. La terapia genetica prevede la possibilità che questo gene possa essere sostituito da uno sano, ossia capace di riportare alla normalità nell’organismo la produzione di emoglobina.
La qualità della vita del paziente talassemico e le complicazioni della talassemia
I pazienti con trasfusioni e chelazioni regolari hanno una buona qualità della vita e possono anche diventare genitori. Mentre fino a trent’anni fa l’aspettativa di vita dei pazienti non superava i 25 anni, oggi la talassemia è una malattia a prognosi aperta!
Tuttavia, le persone affette da Talassemia Major vanno incontro a una serie di complicazioni che derivano dalla malattia e dalle terapie impiegato per la cura dell’anemia mediterranea. Nella scheda vengono elencate alcune delle principali problematiche correlate alla patologia.